Start

During the 1920s a new disease of cattle involving fatal bleeding appeared at about the same time over a wide area of prairie land in north America. It was eventually traced to feeding the animals on hay made from sweet clover (Melilotus alba and M. officinalis) which had 'mysteriously gone bad'. After eating the hay, the animals suffered a gradual decline in the clotting power of the blood (over about 15 days), followed by the development of internal haemorrhage which was generally fatal after 30 - 50 days.
The disease was caused only by feeding the animals on spoilt sweet clover hay, and is commonly called 'sweet clover disease'. It could be treated by transfusing them with blood freshly drawn from normal healthy animals and removing the spoilt hay from the animals' diet.
What conclusions can you draw from these observations?
Obviously there is something in the spoilt sweet clover hay that interferes with the blood clotting process. It is unlikely to be a simple inhibitor, since transfusing the animals with blood from healthy animals restores blood clotting, even though the toxin from the spoilt hay will still be present in their blood.
It is more likely that the toxin interferes in some way with the synthesis of prothrombin, which is activated to thrombin to cause blood clotting.
Roderick (1931) reported on studies of animals with sweet clover disease.
The addition of oxalate or citrate to blood will chelate calcium ions, and prevent clotting. Addition of a solution of calcium chloride to oxalated normal plasma results in rapid clotting; addition of calcium chloride to oxalated plasma from a calf with sweet clover disease did not result in clotting.
What conclusions can you draw from this observation?
Calcium is required for blood clotting - presumably because it binds to prothrombin, but the plasma from the calf with sweet clover disease lacks whatever it is that normally binds calcium to permit clotting. It may be that the affected calf's plasma contains an abnormal prothrombin.
Roderick prepared partially purified prothrombin by precipitation with acetone from blood of either healthy animals or those with sweet clover disease, and tested these two preparations on blood from an affected animal.
| prothrombin prepared from |
healthy calf |
affected calf |
||
| mL prothrombin solution added | 0.5 |
1.0 |
2.5 |
2.5 |
| minutes required for blood to clot | 60 |
45 |
20 |
> 360 |
[From data reported by Roderick LM. A problem in the coagulation of the blood. American Journal of Physiology 96: 413-425 1931]
What conclusions can you draw from this observation?
This experiment shows that it is indeed the prothrombin from the affected animal that is abnormal and inactive. Adding normal prothrombin to the blood of an affected animal permits normal clotting, with more rapid clotting the more prothrombin is added. However, prothrombin prepared from an affected animal does not cause clotting even over 6 hours.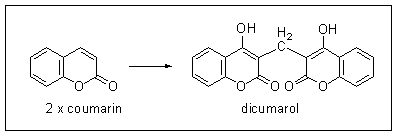
The toxin present in spoiled sweet clover hay was identified as bis-hydroxy-coumarin (dicumarol); it is formed by the oxidation of coumarin which is naturally present in sweet clover.
Once dicumarol was available in adequate amounts, its mode of action could be investigated. Early studies showed that there was a dose-dependent impairment of blood clotting when it was administered to experimental animals, but with a lag period of 12 - 24 hours before any effect became apparent.
What conclusions can you draw from this observation?
Dicumarol is not a simple inhibitor of blood clotting; if it were then the effect would be apparent within a short time of administering it to the animals. The 12 - 24 hour lag period before any effect is observed suggests that it must inhibit the synthesis of normal prothrombin, not simply inhibit its activity. It is obviously not inhibiting the synthesis of something that resembles prothrombin, since Roderick was able to isolate a protein that resembled prothrombin from the blood of an affected animal, but was inactive in initiating blood clotting.
This suggests that dicumarol inhibits some process involve din the formation of active prothrombin from a precursor protein.
Why do you think it was that dicumarol was adopted for use in low doses to reduce blood clotting in patients at risk of thrombosis and in relatively high doses as a rat poison?
In patients at risk of thrombosis the aim is to reduce the capacity of blood clotting to a safe level. A low dose of dicumarol will reduce the synthesis of active prothrombin and so slow blood clotting.
A high dose of dicumarol will lead to more or less complete inhibition of the synthesis of active prothrombin and when eaten by rats will have the same effect as eating spoilt sweet clover hay had on the affected calves - uncontrolled bleeding, leading to death.
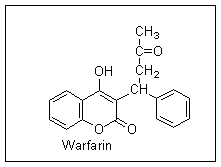
Further research at the University of Wisconsin resulted in the synthesis of a variety of compounds related to dicumarol which were more potent as rat poisons and had fewer side effects in human beings. The most successful of these compounds was called Warfarin (from the initials of the Wisconsin Alumnus Research Fund, who supported the research); it is still the most widely used anticoagulant in clinical use, and one of the most commonly used rodenticides.
Bleeding chickens and a new vitamin
In 1929, Dam and coworkers in Copenhagen were investigating the metabolism of steroids in the chicken. They fed their animals on a fat-free diet, and reported the development of a 'scurvy-like' disease. The animals had subcutaneous and intra-muscular haemorrhages. Other workers reported similar findings when they repeated Dam's studies, and also demonstrated impaired blood clotting in chicks fed on a fat-free diet.
In further studies, Dam et al. showed that the condition was not scurvy (which is due to vitamin C deficiency, and it had already been shown that vitamin C is not a dietary essential for the chicken) and did not respond to vitamin C. They went on to demonstrate a fat-soluble compound in vegetables, grains and animal liver which would normalise blood clotting when fed to deficient chickens.
They proposed the name vitamin K (from the German koagulation) for this new nutrient, and defined a unit of biological activity as that amount required /kg body weight on three successive days to normalise blood clotting in a deficient animal.
[Dam H & Schonheyder F. A deficiency disease in chicks resembling scurvy. Biochemical Journal 28: 1355-1359 1934. Dam H. The antihaemorrhagic vitamin of the chick. Biochemical Journal 29: 1273-1285 1935. Dam H, Schonheyder F & Tage-Hansen E. Studies on the mode of action of vitamin K. Biochemical Journal 30: 1075-1079 1936]
Why do you think the response to vitamin K in deficient animals takes several days to develop?
It is likely that vitamin K is required for the synthesis of one or more of the blood clotting factors (and especially prothrombin). Therefore in deficient animals that lack (active) prothrombin it will take several days to synthesise enough active prothrombin to permit normal blood clotting.
In 1952, Holmes and Love reported on an army recruit who had attempted suicide by taking rat poison containing Warfarin - he consumed some 567 mg of Warfarin over 6 days. On admission to hospital he had numerous subcutaneous haemorrhages and was suffering from nose-bleeds.
His prothrombin time was 54 seconds, compared with a normal value of 14 seconds. He was given 20 mg vitamin K intravenously daily for 10 days, when he recovered, with a normal prothrombin time.
(The prothrombin time is the time taken for the formation of a fibrin clot in citrated plasma after the addition of calcium ions and thromboplastin to activate the extrinsic clotting system).
[Holmes RW & Love J. Suicide attempt with Warfarin, a bishydroxycoumarin-like rodenticide. Journal of the American Medical Association 148: 935-937 1952]
What does this suggest about the way in which Warfarin affects blood clotting?
We have seen that vitamin K is required for the synthesis of (active) prothrombin, and Warfarin inhibits this. It is likely that Warfarin acts as an antagonist of vitamin K.
An abnormal prothrombin after Warfarin or in vitamin K deficiency
Stenflo and coworkers isolated a protein from cattle treated with Warfarin which reacted normally with antiserum to prothrombin, but had no biological activity. During Warfarin treatment, the concentration of this abnormal, inactive, prothrombin in the animals' plasma increased, and that of active prothrombin decreased.
The abnormal prothrombin is less negatively charged than normal prothrombin, and does not migrate so rapidly towards the anode on electrophoresis. The addition of calcium ions to a sample of normal prothrombin reduces its electrophoretic mobility, but has no effect on the electrophoretic mobility of the abnormal prothrombin.
[Stenflo J & Ganrot P-O. Vitamin K and the biosynthesis of prothrombin. Journal of Biological Chemistry 247: 8160-8166 1972]
Nelsestuen and Suttie (1972) showed that normal prothrombin binds 4 mol of calcium /mol of protein, while the abnormal prothrombin from Warfarin-treated animals binds less than 1 mol of calcium /mol.
[Nelsestuen GL & Suttie JW. Mode of action of vitamin K: calcium-binding properties of bovine prothrombin. Biochemistry 11: 4961-4964 1972]
Feeding high intakes of vitamin K to Warfarin-treated animals normalises their blood clotting, and leads to disappearance of the abnormal prothrombin, and reappearance of active prothrombin with normal calcium-binding capacity.
What conclusions can you draw from these results?
The biologically inactive protein that cross-reacted with antiserum to prothrombin is most likely to be a precursor of active prothrombin, which requires modification to gain biological activity. Warfarin inhibits this modification.
The modification of the inactive prothrombin involves introducing negative charges into the protein, and presumably these charged groups bind calcium, since the abnormal inactive prothrombin does not bind calcium.
Shah and Suttie (1974) demonstrated the synthesis of prothrombin in the post-mitochondrial supernatant fraction from liver of vitamin K deficient rats incubated with vitamin K. They demonstrated the formation of prothrombin by its biological activity in a blood clotting assay, and reported their results under various incubation conditions as % of the active prothrombin formed in the presence of 20 µg vitamin K /mL in the incubation:
| incubation conditions | relative activity (%) |
|
1 |
control (20 µg vitamin K /mL) | 100 |
2 |
no vitamin K | 7 ± 1 |
3 |
+ 2-chlorophytylmenaquinone ( an antagonistic vitamin K analogue) + 20 µg vitamin K /mL | 18 ± 1 |
4 |
+ cycloheximide (a protein synthesis inhibitor) + 20 µg vitamin K /mL | 94 ± 1 |
5 |
+ Warfarin + 20 µg vitamin K /mL | 87 ± 3 |
6 |
incubation under anaerobic conditions + 20 µg vitamin K /mL | 0 |
[From data reported by Shah DV + Suttie JW. The vitamin K dependent in vitro production of prothrombin. Biochemical and Biophysical Research Communications 60: 1397-1402 1974]
What conclusions can you draw from these results?
Incubation 2 shows that vitamin K is indeed required for the synthesis of biologically active prothrombin, and incubation 3, containing the antagonistic vitamin K analogue confirms this.
Incubation 4 shows that even when new protein synthesis has been inhibited by cycloheximide, active prothrombin is formed when vitamin K is added to the incubation. This means that there must be a precursor protein already present in the livers of the vitamin K deficient rats, and when vitamin K is added to the incubation this precursor protein can be modified to yield active prothrombin.
Incubation 5 is interesting. We know that Warfarin prevents the formation of active prothrombin, and that an inactive prothrombin-like protein accumulates in Warfarin treated animals, but adding (a relatively large amount of) vitamin K overcomes the effect of Warfarin. This suggests that the antagonistic effect of Warfarin on vitamin K acton is not a simple inhibition. We cannot draw any further conclusions about this without further experiments.
Incubation 6 shows that oxygen is essential for the modification of the precursor protein to active prothrombin, suggesting that an oxidation reaction is involved.
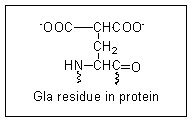 In 1974 Stenflo and coworkers showed that there is a novel amino acid in prothrombin which is not present in the abnormal prothrombin (now called preprothrombin) formed in vitamin K deficiency or on treatment with Warfarin.
In 1974 Stenflo and coworkers showed that there is a novel amino acid in prothrombin which is not present in the abnormal prothrombin (now called preprothrombin) formed in vitamin K deficiency or on treatment with Warfarin.
They identified this amino acid as gamma-carboxyglutamate, (abbreviated to Gla).
There are 10 Gla residues in the amino terminal region of active prothrombin, and these form a binding site for 4 calcium ions.
[Stenflo J, Fernlund P, Egan W & Roepstorff P. Vitamin K dependent modifications of glutamic acid residues in prothrombin. Proceedings of the National Academy of Sciences 71: 2730-2735 1974. Liska DJ & Suttie JW. Location of gamma-carboxyglutamate residues in partially carboxylated prothrombin preparations. Biochemistry 27: 8636-8641 1988.]
All of the codons for amino acids are known.
How does gamma-carboxyglutamate becomes incorporated into prothrombin?
What is the likely role of vitamin K in this process?
Gamma-carboxyglutamate must be formed by modification of glutamate residues in the protein after it has been synthesised - ie glutamate is incorporated during ribosomal protein synthesis, then modified post-synthetically to form Gla.
Vitamin K is presumably a coenzyme or cosubstrate for this post-synthetic modification of glutamate residues in preprothrombin.
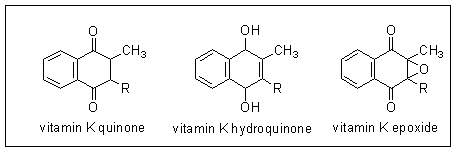 It has been known for some years that vitamin K must be reduced to its hydroquinone for activity in prothrombin synthesis - i.e. it is the hydroquinone which is the active metabolite of the vitamin.
It has been known for some years that vitamin K must be reduced to its hydroquinone for activity in prothrombin synthesis - i.e. it is the hydroquinone which is the active metabolite of the vitamin.
In 1970 Matschiner and coworkers reported that in rats treated with Warfarin and given [14C]vitamin K more than 50% of the radioactivity was found in a new compound, which they identified as vitamin K epoxide. They synthesised the epoxide, and showed that it has the biological activity of vitamin K in deficient animals. Having identified the epoxide, they were able to show that a very small amount is present in the liver of normal animals.
[Matschiner JT, Bell RG, Amelotti JM & Knauer TE. Isolation and characterization of a new metabolite of phylloquinone in the rat. Biochimica et Biophysica Acta 201: 309-315 1970.]
Can you propose the sequence of reactions which vitamin K undergoes in the synthesis of prothrombin?
Which step is likely to be inhibited by Warfarin?
The active form of vitamin K is the hydroquinone, which must be formed by reduction of vitamin K quinone. Vitamin K epoxide is also biologically active, so must be a substrate for reduction to vitamin K hydroquinone, probably via formation of the quinone. The sequence of reactions is shown below.
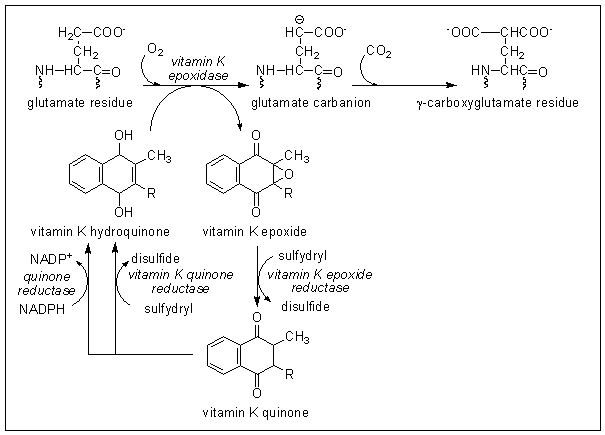
Vitamin K epoxide accumulates in Warfarin-treated animals, suggesting that the action of Warfarin is to inhibit the reduction of the epoxide to vitamin K quinone. This will result in functional vitamin K deficiency in Warfarin-treated animals, since the epoxide cannot be utilised, and will be excreted.
Can you explain why it is that high intakes of vitamin K will overcome the inhibition of prothrombin synthesis caused by Warfarin?
If there is enough vitamin K available when recycling of the epoxide is inhibited by Warfarin then there can be a linear pathway from vitamin K reduction to the hydroquinone, oxidation to the epoxide in the reaction with glutamate and excretion of the epoxide - under these conditions there will be net consumption of vitamin K stoichiometrically with carboxylation of glutamate.
This will also explain the anomalous result in incubation 5 in the experiments by Shah and Suttie (above), who showed that when vitamin K and Warfarin were present together there was little inhibition of prothrombin formation. THere was obviously enough vitamin K present for it to be consumed in the reaction without the need for recycling by reduction of the epoxide.
The classical way of assessing vitamin K nutritional status was by determination of prothrombin time. Can you suggest a more sensitive way of detecting marginal vitamin K deficiency?
Preprothrombin accumulates in the bloodstream in vitamin K deficiency, so if we can raise antibodies against preprothrombin that do not cross-react with prothrombin (ie antibodies that recognise the Gla region of active prothrombin) then we can have a sensitive way of detecting vitamin K deficiency before it is so severe that blood clotting is significantly impaired.
Preprothrombin is sometimes known as PIVKA (Protein Induced by Vitamin K Absence) and assessment of vitamin K status by measuring preprothrombin immunologically is sometimes called PIVKA assay.
Key points from this exercise:
- Vitamin K is required for the synthesis of active prothrombin, which is required for blood clotting.
- Dicumarol and the synthetic compound Warfarin inhibit the synthesis of active prothrombin by inhibiting the normal metabolism of vitamin K.
- Active prothrombin contains a novel amino acid, gamma-carboxyglutamate, which is formed by post-synthetic modification of glutamate residues in a precursor protein, preprothrombin.
- Vitamin K acts as a cosubstrate in the carboxylation of glutamate residues to gamma-carboxyglutamate, and is oxidised to vitamin K epoxide.
- Vitamin K epoxide is normally reduced back to the active vitamin K hydroquinone in a two step reaction; Warfarin inhibits the vitamin K epoxide reductase.
- A high intake of vitamin K will antagonise the anticoagulant action of Warfarin because if there is enough vitamin K quinone present then instead of being recycled, vitamin K epoxide can be excreted, forming a linear rather than cyclic pathway, with net consumption of vitamin K stoichiometrically with carboxylation of glutamate.
- Classically, vitamin K nutritional status was assessed by measuring the prothrombin time (the time taken for the formation of a fibrin clot in citrated plasma after the addition of calcium ions and thromboplastin to activate the extrinsic clotting system), which is still used to assess the efficacy of anticoagulant therapy. A more sensitive way of assessing vitamin K nutritional status is by measuring preprothrombin using specific antibodies that do not cross-react with active prothrombin.
- Preprothrombin is sometimes known as PIVKA (Protein Induced by Vitamin K Absence) and assessment of vitamin K status by measuring preprothrombin immunologically is sometimes called PIVKA assay.
Because batches of thromboplastin vary, manufacturers assign an International Sensitivity Index (ISI) for each batch. The ISI value indicates how that batch of thromboplastin compares with an internationally standardised sample.
The result is then expressed as the Internal Normalised Ratio (INR), which is calculated as the time (in seconds) for the patient's sample to clot divided by the clotting time for a control sample from someone not taking anticoagulants, raised to the power of the ISI.